11. 最局在ワニエ関数¶
ここではquantum-ESPRESSOを用いた電子状態計算から最局在ワニエ関数を構築する方法について紹介します。 そのためのプログラムとして、 wannier90 が公開されています。 このプログラムを利用すると、バンド構造をタイトバインディング模型で表現することができるので、 様々な物理量の計算を簡単に実装したり、k点を細かくとる必要があるような計算の高速化が行えるようになります。 具体的に何ができるようになるかは、wannier90 の web page や 論文 を参考にしてください。
ここでは、wannier90 3.1.0 を用いますが、ここで説明する内容はほとんどバージョンには依らないと思います。
より具体的なマニュアルは、wannier90 の support にある user guide や tutorial を参考にしてください。
注釈
ソースコードの doc/ の中にも user_guide.pdf, tutorial.pdf があります。また、examples/ の中に tutorial.pdf で 説明している例の実際のファイルが含まれています。 ここで説明する以外にも様々な例があり、user_guide.pdf にも非常に詳細な説明がありますので、 詳しくはそちらを参考にしてください。
11.1. インストール¶
Materiapps Live!やUbuntu、WSLでは、
% sudo apt install wannier90
とすればインストールできます。
また、自分でESPRESSOをコンパイルした場合、ESPRESSO のディレクトリの中で
make w90
とやればインストールできます。
Wannier90を自分でコンパイルするなら、以下のように config/ の中の自分の環境にあったファイルをもとに make.inc をつくって、 make してください。
tar zxvf wannier90-3.1.0.tar.gz
cd wannier90-3.1.0
cp config/make.inc.gfort make.inc # gfortran を使う場合
make
11.2. 最局在ワニエ関数の作成¶
ワニエ関数を作成する際には、まずバンドのどの部分を再現するようなタイトバイディング模型を構築するかを考えます。 その際、以下の例で示すように孤立したバンドを再現したい場合と、孤立していない(entangleした)バンドを再現したい場合の2通りのパターンが存在します。 以下では、それぞれの場合について説明していきます。
11.2.1. 孤立したバンドの場合:ダイヤモンドの価電子帯の例¶
まず、孤立したバンドを再現する場合を考えます。 例として、ダイヤモンドの価電子帯のバンド4つに対する最局在ワニエ波動関数を作成してみます。 計算に必要なインプットファイルは以下の4つになります。
最初に、準備として、diamond の scf 計算を行い、次に、6x6x6 のkメッシュにおける電子状態を nscf 計算で求めます。
% pw.x < diamond.scf.in > diamond.scf.out
% pw.x < diamond.nscf.in > diamond.nscf.out
kメッシュのサイズは大きければ大きいほどきれいにバンドをフィットするワニエ関数が得られやすくなります。 ただし、現在のQE+wannier90の実装ではメッシュ状の全てのk点の計算が必要なため、場合によっては計算時間やファイルサイズもかなり大きくなるので注意してください。
注釈
既約なk点のみで計算する方法を開発していますので、興味があれば そちら も参照してみてください。
次に、 wannier90用のインプットファイル( diamond.win )を用いてワニエ関数を作成していきます。 このファイルのそれぞれの値の意味は以下の通りです。
バンドの数、ワニエ関数の数の指定
num_bands = 4 num_wann = 4 exclude_bands=5-8
計算に使用するバンド数(num_bands)と、ワニエ関数の数(num_wann)の指定です。 num_bands は nscf 計算したバンド数(exclude_bands を指定した場合は、nscf 計算でのバンドから exclude_bands で指定したバンドを除いたもの)になります。 今回は、nscf で8バンド計算しているので、exclude_bands で5番目~8番目のバンド(上4つのバンド)を無視します。 num_wann は4つのバンドから4つのワニエ軌道をつくるので4を指定します。
projection の指定
begin projections f= 0.125, 0.125, 0.125: s f=-0.375, 0.125, 0.125: s f= 0.125,-0.375, 0.125: s f= 0.125, 0.125,-0.375: s end projections
projectionsブロックでは、対象とするバンドに対応するワニエ軌道を予想し、それに近い波動関数を指定します。 今の場合、diamond の価電子帯は炭素のsp3混成軌道の結合軌道からなるので、炭素ー炭素のボンド中心にs軌道を置いています。 (f= 0.125, 0.125, 0.125: s は fractional coordinate で (0.125, 0.125, 0.125)の位置に s 軌道を置くという意味になります。) このprojectionの数(4個)は、num_wannと一致していなければなりません。
計算するものの指定
write_hr = .true. bands_plot = .true.
ハミルトニアンの出力、バンドの出力を指定しています。
その他
wannier90 の入力として、quantum ESPRESSO の計算で使用した値をいくつか入力する必要があります。
このファイルを用意した状態で以下のコマンドを実行します。
% wannier90.x -pp diamond
このコマンドは、diamond.win を読み込んで、diamond.nnkp というファイルを作成します。 この diamond.nnkp をもとに ESPRESSO で計算した nscf 計算の内容を wannier90 用に変換します。 変換のためのプログラムは pw2wannier90.x でそのための入力は diamond.pw2wan.in です。
% pw2wannier90.x < diamond.pw2wan.in > diamond.pw2wan.out
このコマンドで、diamond.mmn, diamond.amn, diamond.eig という3つのファイルが作成されます。 この3つのファイルと diamond.win を使って、最後に最局在ワニエ波動関数を作成します。
% wannier90.x diamond
これで、diamond_band.{gnu,dat,kpt} というワニエで作成したバンドの情報、 diamond_hr.dat というワニエ関数をベースとして表現したハミルトニアン(タイトバインディング模型)が作成されます。
作成したバンドは gnuplot で
% gnuplot
gnuplot> load 'diamond_band.gnu'
としてやればみることができます。 もとのESPRESSOで計算したバンドとの比較は以下のようになります。黒がDFTの結果で、赤がwannier90で作成したタイトバイディング模型のバンドです。
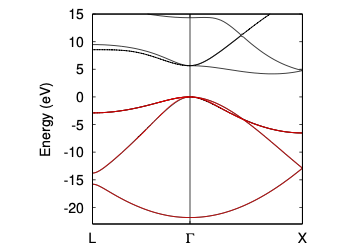
また、得られたワニエ軌道の中心や広がり(spread)は dimond.wout の中で以下のように出力されます。
Final State WF centre and spread 1 ( -0.440540, 0.440540, 0.440540 ) 0.72903198 WF centre and spread 2 ( 0.440540, 0.440540, -0.440540 ) 0.72903199 WF centre and spread 3 ( -0.440540, -0.440540, -0.440540 ) 0.72903199 WF centre and spread 4 ( 0.440540, -0.440540, 0.440540 ) 0.72903199 Sum of centres and spreads ( -0.000000, -0.000000, -0.000000 ) 2.91612794
これは例えば1番目のワニエ軌道の中心が Cartesian coordinate Angstrom単位で (-0.440540, 0.440540, 0.440540)、ワニエ軌道の広がり(spread)が、0.72903198 (Angstrom^2) ということを示しています。 これをみれば、4つ等価な軌道がボンド中心にちゃんと現れていることが確認できます。
11.2.2. entangleしたバンドの場合:グラフェンのsp2 と π軌道の例¶
一般には、モデル化したいバンドが孤立していることは珍しく、たいていの場合、他のバンドと重なりがあります。 そこで次に、このようなバンドがentangleした場合の例としてグラフェンの価電子帯における sp2軌道とFermi準位前後にあるπ軌道に対する最局在ワニエ波動関数を作成してみます。 必要なファイルは以下の通りです。
まず、準備として、graphene の scf 計算を行い、12x12x1 のメッシュにおける電子状態を nscf 計算で求めます。 nscf 計算では、nbnd オプションで計算するバンド数を指定していることに注意してください。
% pw.x < graphene.scf.in > graphene.scf.out
% pw.x < graphene.nscf.in > graphene.nscf.out
次に、 wannier90用のインプットファイル( graphene.win )を用いてワニエ関数を作成していきます。 このファイルのそれぞれの値の意味は以下の通りです。
バンドの数、ワニエ関数の数の指定
num_bands = 16 num_wann = 5
今回は、16本のバンドの中から5個のワニエ軌道からなるバンドを抜き出します。16はnscf計算で指定したバンド数になります。
Outer window, Inner window の指定
dis_win_min = -30 dis_win_max = 12 dis_froz_min = -30 dis_froz_max = 2.6
Inner window (dis_froz_max,min) はワニエを作る際、ワニエ軌道によるバンドが完全にもとのバンドと一致するべき範囲で、Outer window (dis_win_max,win) はワニエを作る際に考慮するバンドが含まれている範囲を指定します。(下図参照)
今の場合、E = 2.6 eV 以下のバンドは全て sp2軌道およびπ軌道で表されるべきバンドなので、E<2.6eVをInner window に指定しています。 逆にΓ点の E=2.6 eVより少し上にあるバンドは、sp2やπ軌道由来のものではないので、Inner window にこのバンドを含めてしまうとワニエがうまく作れなくなってしまいます。 一方、π軌道の存在する範囲は上にE ~ 12eV近くまであるため、Outer window はこのπ軌道を全て含むように E<12eVで指定しています。 これらの値は、別途バンド計算などをして確認する必要があります。 このページの下の方にあるDFTとワニエのバンドの比較も参照してみてください。
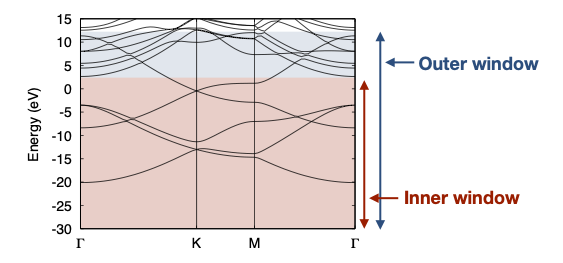
projection の指定
begin projections C: pz f= 0.167, 0.333, 0.00: s f=-0.333,-0.167, 0.00: s f= 0.167,-0.167, 0.00: s end projections
ワニエ関数の初期値として炭素上のπ軌道(pz)とC-Cボンドの中心のs軌道(sp2 混成軌道の結合軌道に対応)を指定しています。 C: pz は炭素のある位置全てに pz を置くことを意味していますので、この一行で2つprojectionを指定したことになります。 f で始まる3行で sp2 の結合軌道を表しており、π軌道と合わせて全部で5個のprojectionを指定したことになります。
計算するものの指定
write_hr = .true. bands_plot = .true. wannier_plot = .true.
ハミルトニアンの出力(write_hr)、バンドの出力(bands_plot)の他に、今回はワニエ波動関数の出力(wannier_plot)を指定しています。 ワニエ関数の出力をするには、pw2wannier.x のインプットで write_unk = .true. にしておく必要があります。
その他
wannier90 の入力として、quantum ESPRESSO の計算で使用した値をいくつか入力する必要があります。
このファイル(graphene.win)を用いて graphene.nnkp を作成します。
% wannier90.x -pp graphene
これをもとに ESPRESSO で計算した nscf 計算の内容を wannier90 用に変換します。
% pw2wannier90.x < graphene.pw2wan.in > graphene.pw2wan.out
今回は、ワニエ波動関数を作成するために、write_unk = .true. としたため、graphene.{mmn,amn,eig} の他にUNKで始まるファイルが出力されます。
最後に最局在ワニエ波動関数を作成します。
% wannier90.x graphene
これで、graphene_band.{gnu,dat,kpt} というワニエで作成したバンドの情報、 graphene_hr.dat というワニエ関数をベースとして表現したハミルトニアン(タイトバインディング模型)、 5つの最局在ワニエ波動関数 graphene_00001.xsf - graphene_00005.xsf が生成されます。
まず、graphene.wout の出力を確認すると、
Final State WF centre and spread 1 ( 0.000000, 0.000000, -0.000000 ) 0.95935107 WF centre and spread 2 ( -0.000000, 1.406006, -0.000000 ) 0.95935091 WF centre and spread 3 ( 0.000000, 0.703003, 0.000000 ) 0.60491046 WF centre and spread 4 ( -0.608818, -0.351502, 0.000000 ) 0.60491046 WF centre and spread 5 ( 0.608818, -0.351501, -0.000000 ) 0.60491046 Sum of centres and spreads ( 0.000000, 1.406006, -0.000000 ) 3.73343336
のように、1,2番目のワニエ軌道が炭素位置にあるπ軌道、3,4,5番目がボンド中心にあるsp2結合軌道になっていることがわかります。
ワニエ関数の具体的な形は graphene_00001.xsf - graphene_00005.xsf を XCrysden や VESTA で開くことで確認できます。 実際1番目のワニエ軌道 graphene_00001.xsf を XCrysden でプロットすれば、得られた最局在ワニエ関数を以下のようにプロットでき、確かにπ軌道が得られていることが分かります。
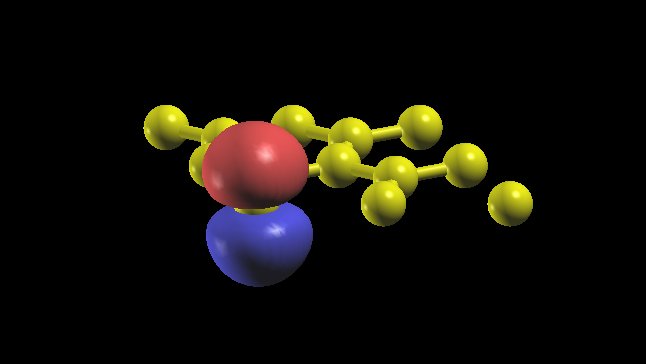
ワニエで作成したバンドは gnuplot でプロットできます。
% gnuplot
gnuplot> load 'graphene_band.gnu'
もとのESPRESSOで計算したバンドとの比較は以下のようになります。(黒:DFT、赤:ワニエ)

最後に、graphene_hr.dat ファイルをみてみます。 このファイルは、transfer integral のリストが出力されており、それぞれの行が1つの transfer integral の値になります。 各行が、
a b c m n Re(t) Im(t)
という形になっており、m 番目のワニエ軌道から(a,b,c)の位置にある単位胞のn番目のワニエ軌道へのt の値を意味します。 例えば、
0 0 0 5 1 0.000000 -0.000000
0 0 0 1 2 -3.001042 0.000000
0 0 0 2 2 -0.154412 0.000000
0 0 0 3 2 -0.000000 0.000000
0 0 0 4 2 -0.000000 -0.000000
...
ではハイライトした行が、1 番目のワニエ軌道((0.0, 0.0, 0.0)にあるπ軌道)から同じ単位胞内の2番目のワニエ軌道((0.0, 1.406, 0.0)にあるπ軌道) へのホッピング(Re(t) = -3.001042, Im(t) = 0.000000)を表し、
0 0 0 5 5 -11.355711 0.000000
0 1 0 1 1 0.231742 -0.000000
0 1 0 2 1 -3.001042 -0.000000
0 1 0 3 1 -0.000000 -0.000000
0 1 0 4 1 0.000000 -0.000000
...
では、ハイライトした行が、1 番目のワニエ軌道((0.0, 0.0, 0.0)にあるπ軌道)から(0,1,0)の位置にある単位胞の1番目のワニエ軌道(a_2 + (0.0, 0.0, 0.0)にあるπ軌道) へのホッピング(Re(t) = -0.231742, Im(t) = 0.000000)を表します。
したがって、graphene_hr.dat ファイルをみると最近接、次近接の transfer integral がそれぞれ t = -3.00 eV, t'=0.23 eV と得られている、といったことが分かります。
11.2.3. entangleしたバンドの場合:スピン軌道相互作用がある強磁性Feの例¶
最後にスピン軌道相互作用がある場合の例として Fe の強磁性状態を考えます。 ワニエ軌道としては、Fe の原子軌道(3d,4s,4p)を考えます。
Feの擬ポテンシャルとして Fe.rel-pbe-spn-rrkjus_psl.0.2.1.UPF を使いますので、同名のファイルを公式サイトからダウンロードしてください。
注釈
なお、このウルトラソフト擬ポテンシャルでスピン軌道相互作用があるとき、あるいはノンコリニアな磁性があるときのワニエ関数をつくるにはespresso のバージョン 6.0 以上が必要となります。この機能を使う場合、wannier90-3.0.0の論文 を引用してください。
必要なファイルは以下の通りです。
スピン軌道相互作用の入った scf 計算をするには、スピン軌道相互作用の入った擬ポテンシャルを用意し Fe.scf.in , Fe.nscf.in に 以下のオプションを追加します。
degauss = 0.02
lspinorb = .true.
まず、電子状態を計算します。 nscf 計算では、ワニエ関数作成に使うバンドの数も指定しておきます。(nbnd = 68)
% pw.x < Fe.scf.in > Fe.scf.out
% pw.x < Fe.nscf.in > Fe.nscf.out
次に、 wannier90用のインプットファイル( Fe.win )をみていきます。
バンドの数、ワニエ関数の数の指定
num_bands = 60 num_wann = 18 exclude_bands = 1-8
nscf 計算で68バンド計算しましたが、下の8バンドはFeの3s,3pからなるバンドでかなり深い位置にあるので、除いています。残りの60本のバンドから、18個のワニエ軌道を作ります。
iteration の回数指定
dis_num_iter = 200 num_iter = 0
dis_num_iter は disentangle するときの iteration の回数を、 num_iter は disentangle した後の最局在化の iteration の回数を指定しています。 num_iter = 0 とすると、最局在化は行いませんが、そのかわり projection で指定した波動関数の対称性をほぼ保ったワニエ軌道が得られやすくなります。
spinors オプション
スピン軌道相互作用がある場合、ノンコリニアな磁性を扱う場合など、波動関数がスピノルで表現される場合は、spinors オプションを追加します。
spinors = .true.
projection の指定
begin projections Fe:sp3d2;dxy;dxz;dyz end projections
Feの3d,4s,4pからなる軌道として、sp3d2,dxy,dyz,dzx の軌道を指定しています。 spinors オプションを指定しているため、これで(1+3+5)*2=18個のワニエ軌道を指定したことになります。
注釈
qe-7.2 以上であれば単純に、Fe: s,p,d としてもうまくいきます。 もし、ワニエ関数が微妙にうまく作れない、といった問題があるときは、qe-7.2以上のpw2wannier90.xを使ってみてください。
あとはグラフェンの場合と同様に
% wannier90.x -pp Fe
% pw2wannier90.x < Fe.pw2wan.in > Fe.pw2wan.out
% wannier90.x Fe
とやればワニエ関数が作れます。
Fe.wout で得られたワニエ軌道を確認してみると以下のようになっています。
Final State
WF centre and spread 1 ( -0.535119, 0.000000, -0.000000 ) 0.61515070
WF centre and spread 2 ( -0.609202, 0.000000, -0.000000 ) 0.58968845
WF centre and spread 3 ( 0.535119, 0.000000, -0.000000 ) 0.61515072
WF centre and spread 4 ( 0.609202, 0.000000, 0.000000 ) 0.58968841
WF centre and spread 5 ( 0.000000, -0.535119, 0.000000 ) 0.61515070
WF centre and spread 6 ( 0.000000, -0.609202, 0.000000 ) 0.58968841
WF centre and spread 7 ( 0.000000, 0.535119, 0.000000 ) 0.61515073
WF centre and spread 8 ( -0.000000, 0.609202, -0.000000 ) 0.58968847
WF centre and spread 9 ( 0.000000, 0.000000, -0.534873 ) 0.61421779
WF centre and spread 10 ( -0.000000, 0.000000, -0.609034 ) 0.58986115
WF centre and spread 11 ( -0.000000, -0.000000, 0.534873 ) 0.61421775
WF centre and spread 12 ( 0.000000, -0.000000, 0.609034 ) 0.58986112
WF centre and spread 13 ( 0.000000, 0.000000, 0.000000 ) 0.45083453
WF centre and spread 14 ( 0.000000, 0.000000, 0.000000 ) 0.49145010
WF centre and spread 15 ( 0.000000, 0.000000, -0.000000 ) 0.45083453
WF centre and spread 16 ( -0.000000, 0.000000, 0.000000 ) 0.49145005
WF centre and spread 17 ( -0.000000, -0.000000, -0.000000 ) 0.45093816
WF centre and spread 18 ( 0.000000, -0.000000, -0.000000 ) 0.49140832
Sum of centres and spreads ( 0.000000, -0.000000, 0.000000 ) 10.05443008
最初の12個がsp3d2の軌道で、残りが、dxy, dyz, dzx になっています。 それぞれの軌道の up, down が交互に並んでおり、dxyとdyzは同じサイズのワニエ軌道ができている、といったことが分かります。
11.3. wannier90 を使うときの注意点¶
結果を確認するときは、まず wannier90 でできたバンドと wout ファイルに記述されている各ワニエの spread を見ます。 バンドが波打っていたり、spread が大きい場合はワニエの作成に失敗していますので、パラメータを確認してください。 spread の典型的な大きさは、s軌道,p軌道では1~4 A^2、d軌道では1 A^2 以下になります。
うまく作れないときは、たいていfitするために選ぶバンドと projection で指定する最初の波動関数の予想が合っていないことが原因です。 winファイルで、exclude_bands, dis_win_min, dis_win_max, dis_froz_min, dis_froz_max, projections をうまく設定してください。
quantum ESPRESSO では擬ポテンシャルを用いているので、計算の対象となるバンドは擬ポテンシャルに依存します。(Fe の例では、nscf 計算で 3s, 3p を含んだバンドが得られていますが、3s, 3p を含まない擬ポテンシャルも存在し、その場合は 3s, 3p を含まないバンドが得られます。)各元素で考慮している軌道、およびそのおおよそのエネルギーは擬ポテンシャルのファイルに記述されていますのでその値を参考にしてください。 このようなことを自動的に考慮して、自動的に結晶構造ファイル(cifファイル)から、quantum ESPRESSOとwannier90のインプットを作成するスクリプトとして、 cif2qewan を開発していますので、興味があればそちらも参照してみてください。